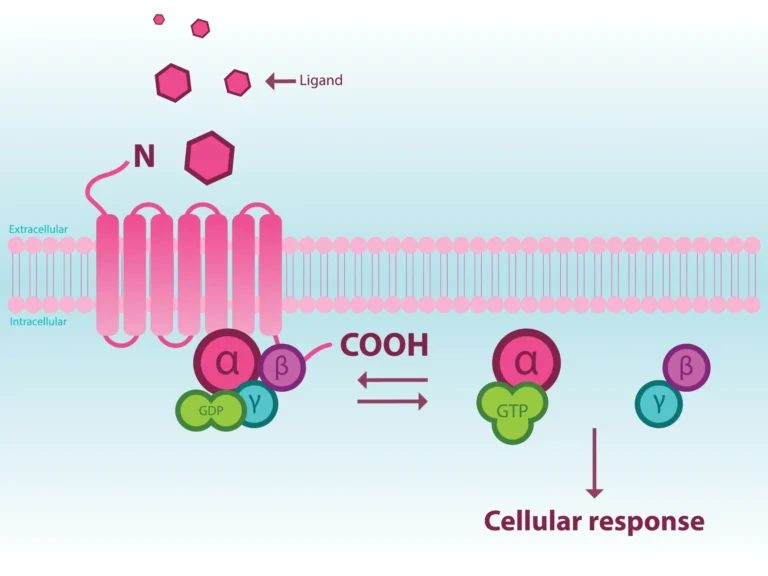
G protein-couple receptors (GPCRs) are an extremely diverse group of membrane receptors that play vital roles in a variety of normal and disease-associated pathologies and remain key targets in drug development. As such, researchers continue to scrutinize the complex pathways of GPCR activation and their regulation of downstream cellular and physiological responses. And with each new wave of GPCR knowledge comes new avenues for therapeutic intervention. Continued advancements in GPCR research and clinical translation of these novel findings, however, depends on the toolbox of available assays. As our understanding of GPCRs evolves, so to must our cell-based assays evolve.
A Focus on β-arrestin
One area of rapidly expanding knowledge is the role β-arrestins play in mediating GPCR signaling. As the name suggests, GPCRs were originally thought to initiate signal transduction via associated G proteins, while β-arrestin was believed to play a more inhibitory role involved in the desensitization of GPCRs.
Subsequent research has shown that β-arrestins, both in conjunction with and independently of G proteins, mediate a number of ligand activated GPCR activities including receptor internalization, trafficking, and receptor desensitization as well as initiation of signaling via distinct kinase cascades including MAPK and PI3K. One commonality is that all β-arrestin-mediated activities begin with translocation of β-arrestin to the activated GPCR.
Upon activation, GPCRs interact with three main families of proteins – G proteins, GPCR kinases (GRKs), and arrestins. These interactions result in bimodal GPCR activation: G protein-mediated activation and/or GRK and β-arrestin-dependent activation. Ligands may stimulate both G protein- and β-arrestin- mediated modes of signaling or preferentially use one mode over another, an effect known as biased signaling.
Studies have revealed that GPCR conformational changes and GRK phosphorylation patterns upon ligand binding are responsible not only for initiating cellular signaling, but also for determining which signaling mode(s) is stimulated. One means of regulating signaling pathway usage is via the recruitment and differential phosphorylation of the carboxy-terminal tail of ligand-activated GPCRs by different GRK isoforms. The resulting pattern of phosphorylation, often referred to as the phosphorylation ‘barcode’, mediates subsequent cellular outcomes, including β-arrestin involvement.
Exploiting GPCR Bias
Since discovering the bimodal nature of GPCR signaling, ligand-dependent biased signaling has been described. Moreover, research has demonstrated that altering a GPCR’s biased signaling can lead to divergent cellular outcomes. This highlights the need to better characterize both G protein- and β-arrestin-mediated signaling not only of natural ligands but also the effects of candidate drugs. These insights may ultimately produce more efficacious drugs with improved safety profiles and fewer undesired side effects.
Assessing biased ligand activity requires the ability to independently monitor G protein- and β-arrestin-mediated signaling. With the expanding importance of β-arrestins in GPCR physiology, robust β-arrestin cell-based assays have become a critical piece of GPCR-targeted drug development.
A variety of cell-based GPCR G-protein signaling assays are available in formats suitable for high throughout screening in drug development campaigns. In contrast, a limited number of β-arrestin assays are available.
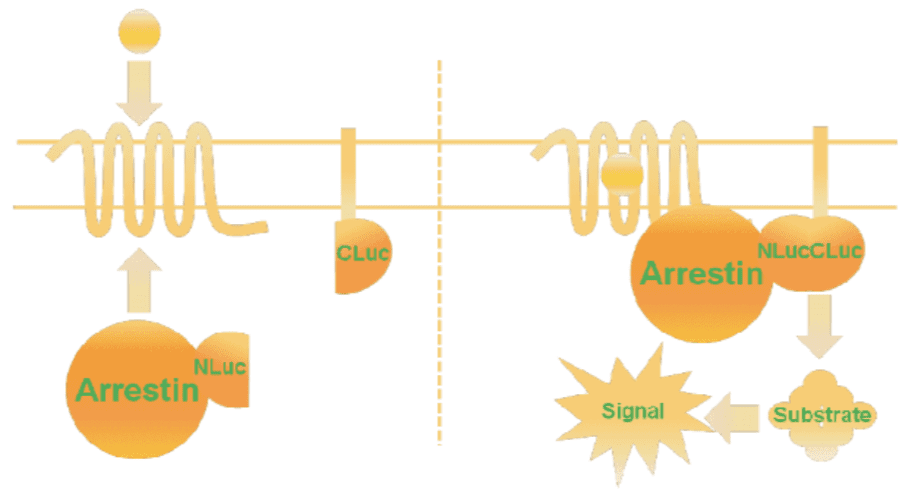
Current cell-based assays use arrestin translocation as the read out for stimulation of β-arrestin. First generation assays rely on tagging both the GPCR-of-interest and β-arrestin with proteins that when brought together mediate production of light.
GPCR tagging can be deleterious, induce receptor conformational changes, lead to steric hinderance, or modify receptor pharmacology – all of which impact the identification and optimization of highly qualified drug candidates. Moreover, these assays require the overexpression of the tagged GPCR, precluding the assessment of native, endogenously expressed GPCR targets.
Freeing Your GPCR - The Next Generation
The MULTISCREEN™ β-Arrestin Sensor technology uses translocation of β-arrestin as the assay readout; however, it is β-arrestin and a membrane ‘sensor’ that are tagged. Upon recruitment, tagged arrestin comes within proximity of a membrane sensor which in turns results in a functional luciferase enzyme. This scheme overcomes receptor tagging drawbacks and importantly offers a means to examine β-arrestin activation via endogenous or orphan GPCRs using an assay format well suited for high throughput, cell-based screening.
Why Set Your GPCR Free?
- To assess GPCRs in their native form for true pharmacology
- To assay endogeously expressed GPCRs for more relevant data
- To characterize orphan GPCRs to expand your target pool
- To enable use of a single cell line for multiple GPCR assays for accelerated drug development
The Ultimate β-Arrestin Partner
Whether lacking specific areas of expertise, being constrained by resources, or facing compressed project timelines, Multispan realizes that each client faces different challenges in their GPCR drug development efforts. We have tailored our solutions for β-arrestin assays, including:
- MultiScreen™ β-Arrestin reagent options
- HSV and Bacmam viral particles carrying MultiScreen™ β-Arrestin sensor genes
- β-Arrestin sensors plasmids
- MultiScreen™ β-Arrestin Assay Kits (Catalog MSBAK01)
- MultiScreen™ β-Arrestin BacMam or HSV Assay Kits (Catalog MSBAKBM01-1, MSBAKHSV01-1)
- Cell Lines
- > 1000 GPCR-expressing cell lines
- 20+ GPCR stable cell lines co-expressing β-Arrestin sensor
- HEK293 and CHO parental cell lines stably expressing MultiScreen™ β-Arrestin sensors
- Services
- Custom stable cell line generation
- MultiScreen™ β-Arrestin functional assay development
- GPCR screening and profiling services using β-Arrestin assays
- Ready-to-use, division-arrested cells
For more information on β-arrestin-mediated signaling, check out this review: G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. J Cardiovasc Pharmacol. 2017 Sep;70(3):142-158.